NMRPredict Help
| Help overview | ||
| Running a Prediction | ||
| Inspecting Prediction Results | ||
| C13 NMR Prediction in Detail | ||
| Proton NMR Prediction in Detail |
Setting Proton Prediction Options
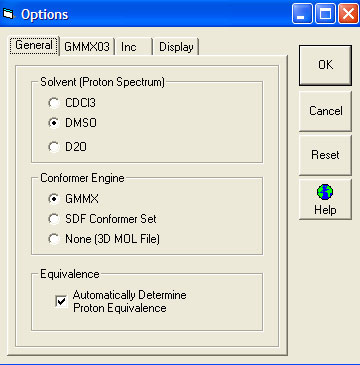
General Tab
- Solvent (Proton Spectrum)
- CDCl3 - use functional groups parametrised in CDCl3
- DMSO – use functional groups parametrised in DMSO
- D2O – use functional groups parametrised in D2O
- Conformer Engine
- GMMX - This is the default setting. GMMX will be called usisng the settings above.
- SDF Conformer Set GMMX will not be called. You will be providing the program with an SD file containing multiple 3D structures. Note that all hydrogens need to be explicitly drawn. The SD file will be passed directly to CHARGE for proton prediction.
- None (3D MOL file) You will be providing the program with a 3D MOL file. Note that all hydrogens need to be explicitly drawn. The MOL file will be passed directly to CHARGE for proton prediction.
- Equivalence
- Automatic - the default is to have this box clicked which will automatically recognise all equivalent carbon atoms and set them as being equivalent.
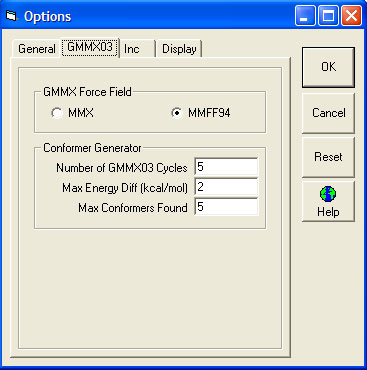
- GMMX Force Field
- MMX
- MMFF94
- Conformer Generator
- Number of GMMX03 Cycles The default is 5.
- Max Energy Diff (kcal/mol) - Determines whether or not a conformer is passed to the CHARGE program for prediction, and thus is included in the averaging process. Conformers whose energy is greater than this value above the minimum energy are excluded. The default is 2.
- Max Conformers Found The default is 5.
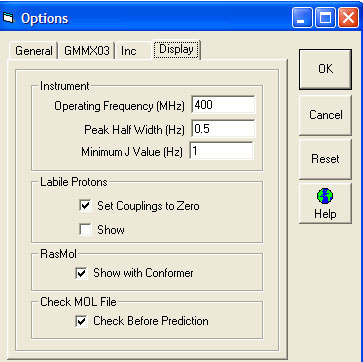
- Instrument
- Operating Frequency (MHz) -
- Peak Half Width (Hz) –
- Minimum J Value (Hz) –
- Labile Protons
- Set Coupling to Zero - the default is to have this box clicked and to set the couplings for all labile protons to zero
- Show - the default is not to show predicted labile protons
- RasMol
- You can choose here whether you want to see the 3D molecules in RasMol as you step through the conformers.
- Check MOL file
- The default is for NMRPredict to attempt to clean the molecule before it is sent for prediction. This helps when users mistakenly draw invalid structures.
GMMX03 Tab
The 2D or 2.5D MOL file representation of the structure is converted by GMMX into a 3D structure and then this structrure is repeatedly minimised to produce a series of conformers. The parameters "Number of GMMX03 Cycles" and "Max Conformers Found" affect the number of conformers produced by GMMX. There is, however, no simple relationship between these values and the actual number of conformers.
Inc Tab
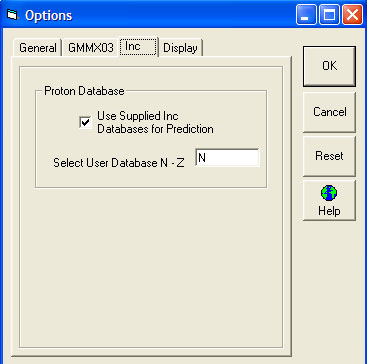
The default here is to click the box saying "Use Supplied Inc Databases for Prediction". This is database "A" which has been hand selected by Professor Pretsch to compliment his standard increment predictions. It is not recommended to turn this off.
If you have created user databases (N to Z) these are selected in the available box.
Display Tab
These parameters are used to generate the proton spectrum. The 'Minimum J Value (Coupling Constant) is required since numerical calculation of coupling constants can lead to many small unobservable couplings which make the display of the spectrum extremely slow.