| Home | Products | Consulting Services | Contact us |
NMRPredict:
| Modgraph Home | ||
| NMRPredict Overview | ||
| Proton NMR Prediction Overview |
HOSE code and Substituent Chemical Shift Prediction
Professor Ernö Pretsch of ETH, Zürich is the founder of the substituent chemical shift approach for proton NMR prediction and we are delighted to have it included in NMRPredict.
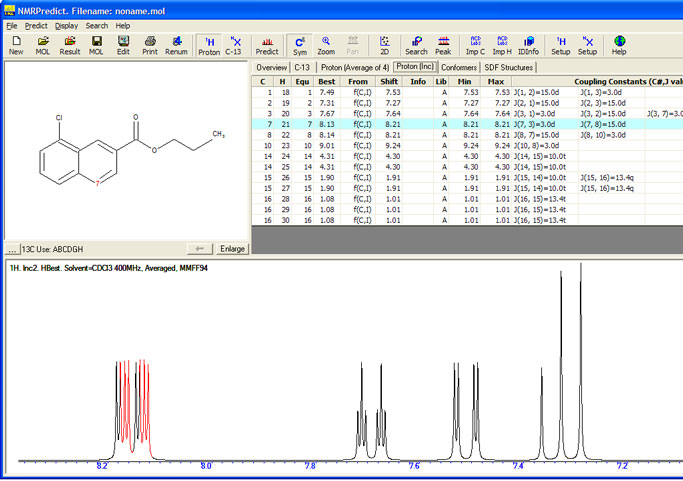
We now have 27,947 recent proton NMR spectra abstracted from the literature behind our proton predictions. When we are unable to make a prediction at two "shells" from the HOSE code database we use the substituent chemical shift approach.
Prediction of 1H-NMR shift values are based almost 3000 parameters, chemical shifts are predicted using additivity rules and several strategies of approximation.Chemical shifts are predicted for all atoms for which additivity rules are available. For a given molecule, the appropriate substructures are automatically assigned following a hierarchical list. These substructures provide the base value of a final predicted shift. Ring systems not available in the data set are approximated by embedded rings and, if necessary, even disassembled into acyclic substructures.
For each substructure found, the remaining parts of the molecule are treated as substituents. Substituents contribute to a final shift by its increments added to the base value. If increments for those substituents are not available so-called embedded substituents, smaller structural units with the same neighbouring atoms, are applied automatically. Or increments of identical or embedded substituents of a corresponding substructure are taken assuming that the effects of the substituents are of the same magnitude.
Models for ethylenes (cis/trans) and cyclohexanes (eq/ax) are also implemented. To use this feature, cyclohexanes must be drawn in the chair conformation